RXN for Chemistry - Predict retrosynthesis
This guide is a tutorial to learn how to predict a retrosynthesis using RXN for Chemistry.
Start Tutorial
| An RXN for Chemistry account is required to complete this tutorial. |
|
Step 1: Log into RXN for Chemistry
| See our RXN for Chemistry Quick Start for directions on logging into the RXN for Chemistry system. |
Step 2: From the RXN for Chemistry Home screen, click "Predict retrosynthetic routes"
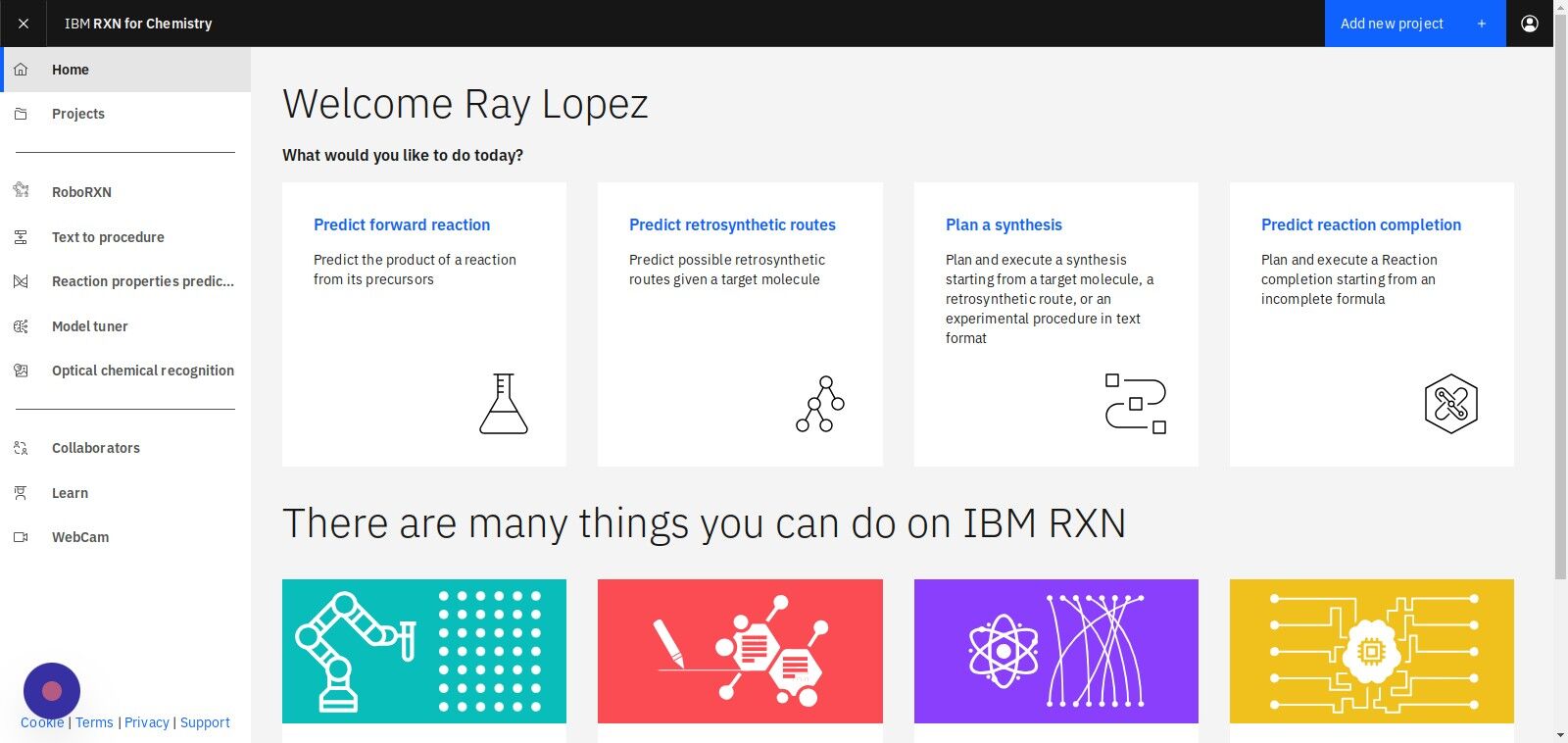
Step 3: Select "Add to pre-existing project" or "Add to a brand new project"
If selecting Add to pre-existing project then use the dropdown labeled "Filter" to select the project.
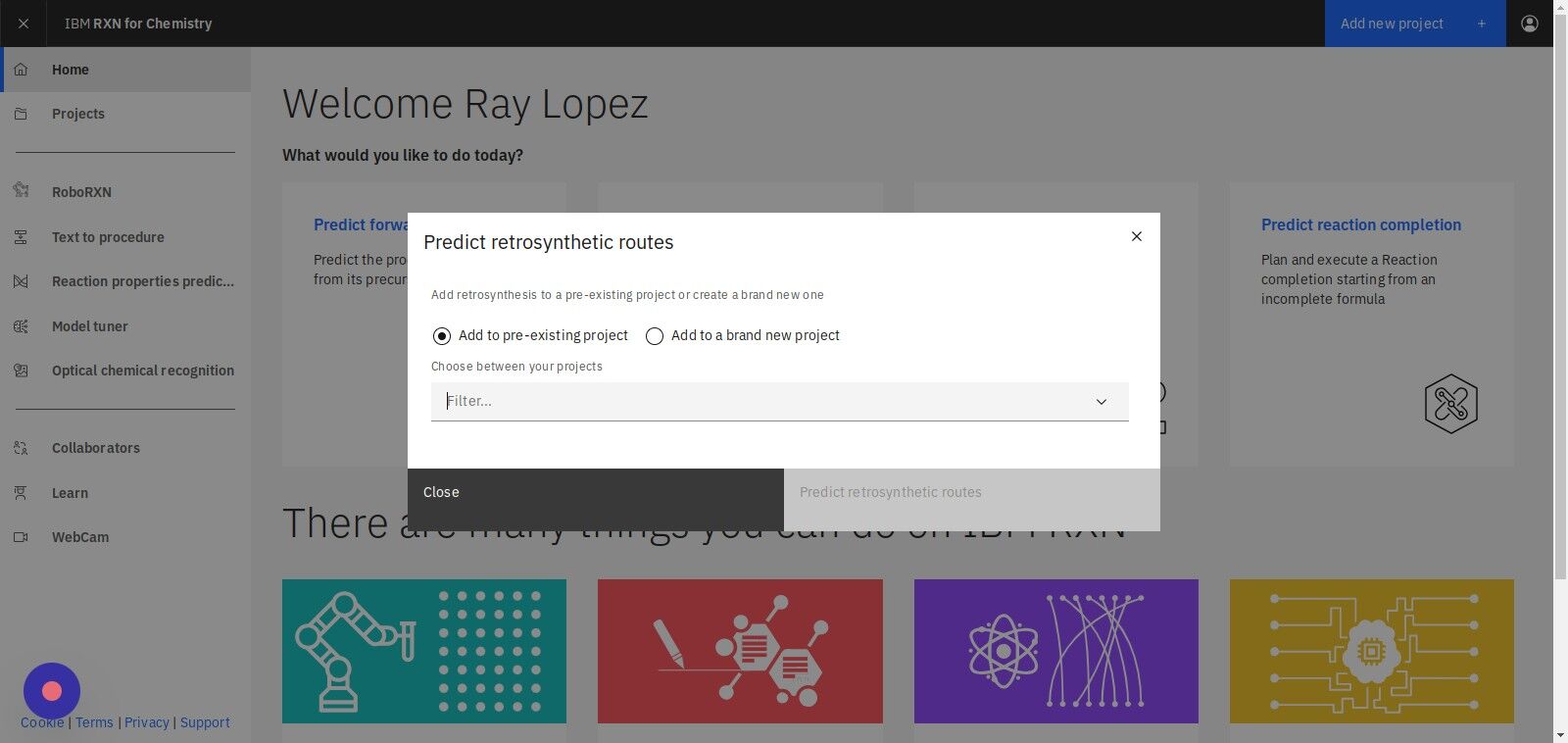
Step 4: Select "Target molecule" to specify a molecule
| If you have a file containing SMILES strings you may use the "From file" option to upload that file. |
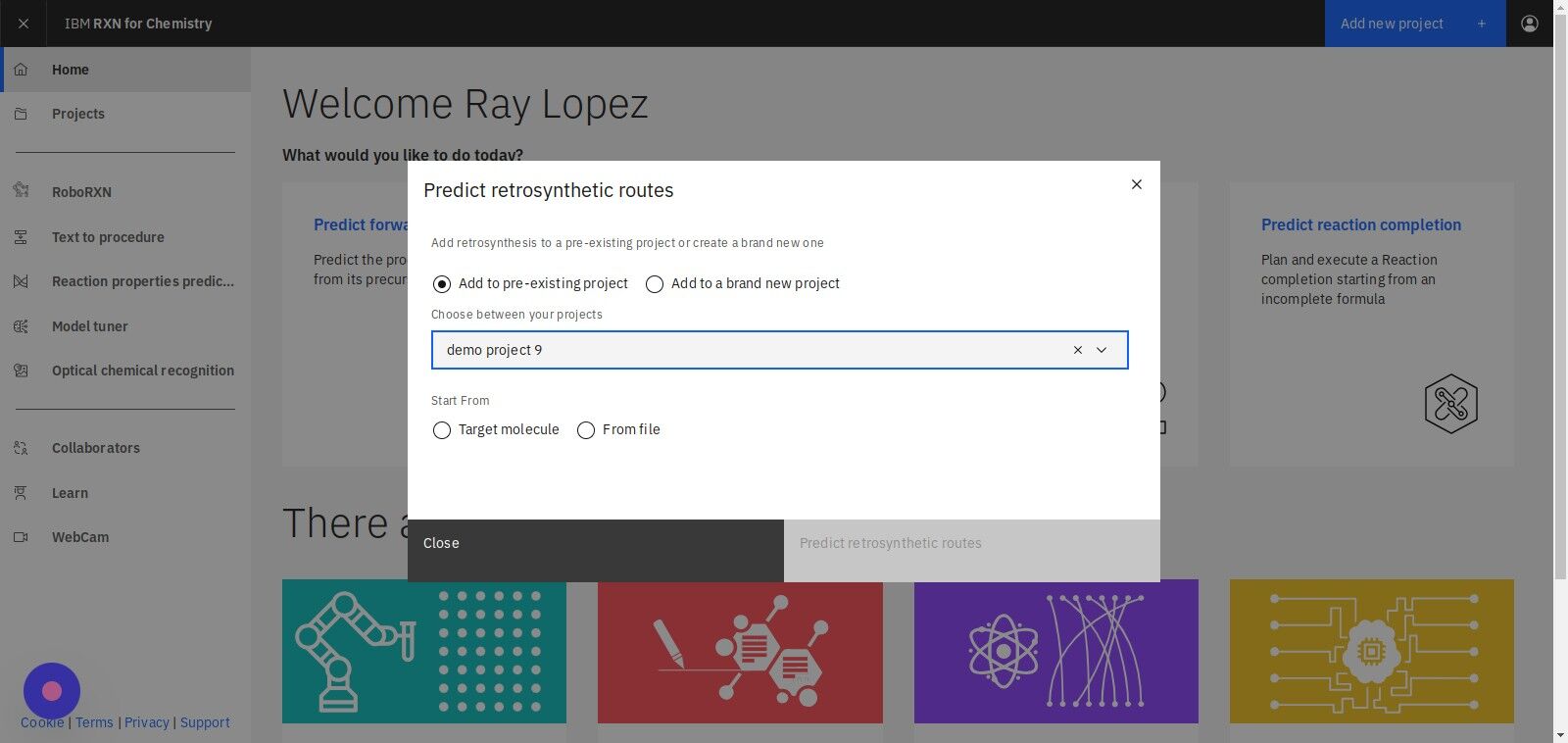
Step 5: Click "Predict retrosynthetic routes"
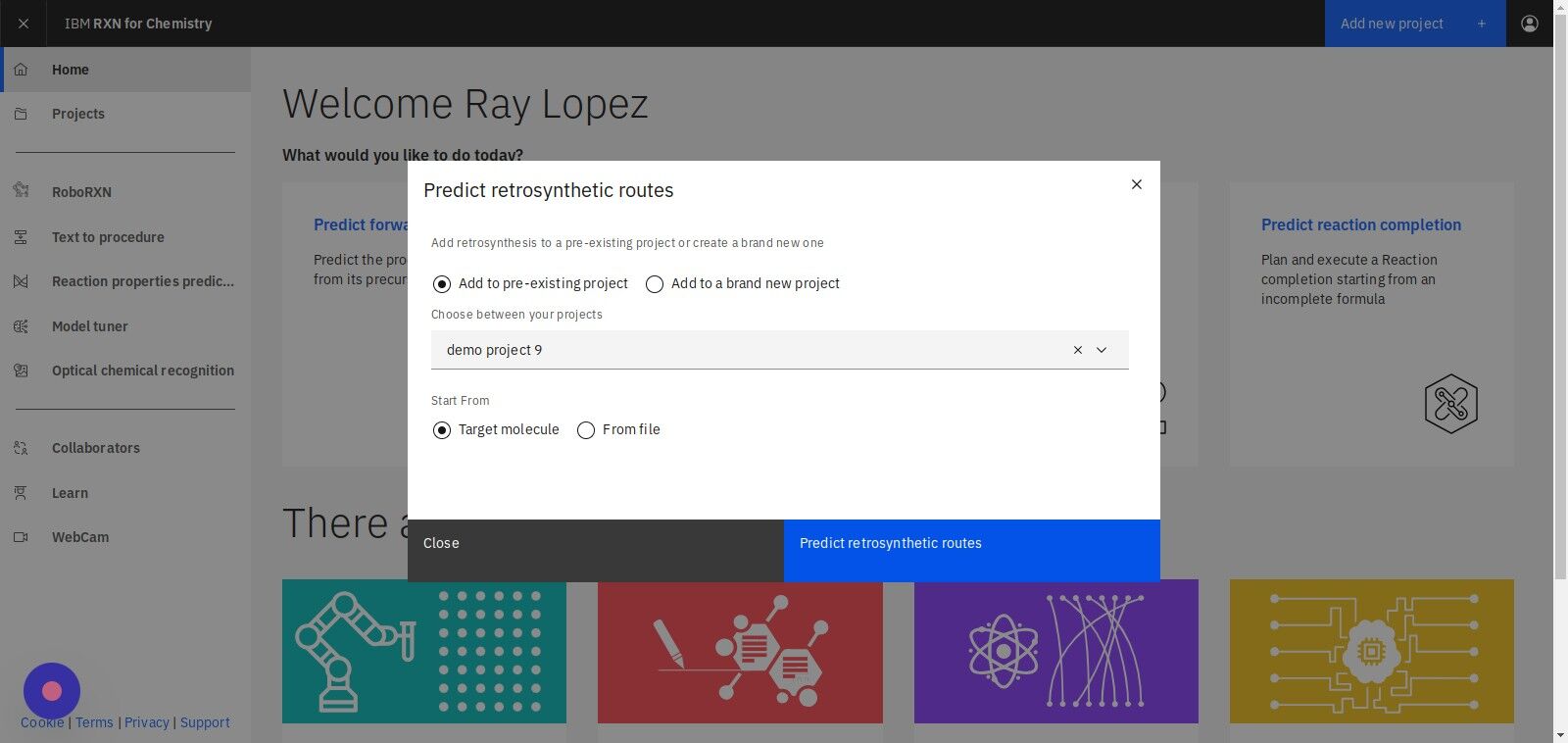
Step 6: In the visual editor, click "Smiles string editor" at the top of the page
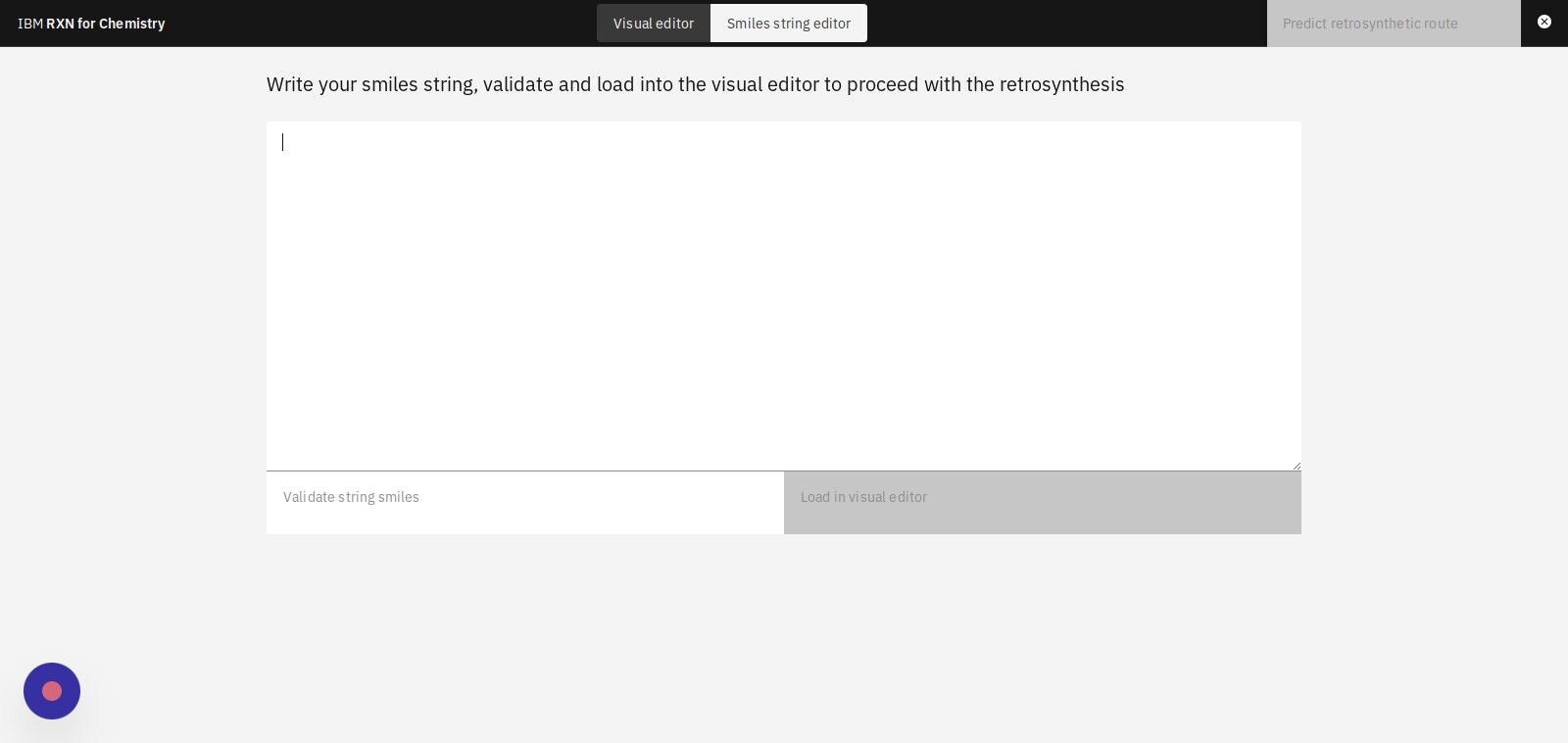
Step 7: Copy and paste a SMILES string into the text box.
On this page, you will paste a SMILES string representing the "target" molecule for the retrosynthesis. Use the one shown below.
The following SMILES string is for a molecule named paliperidone palmitate (brand name Invega Sustenna).
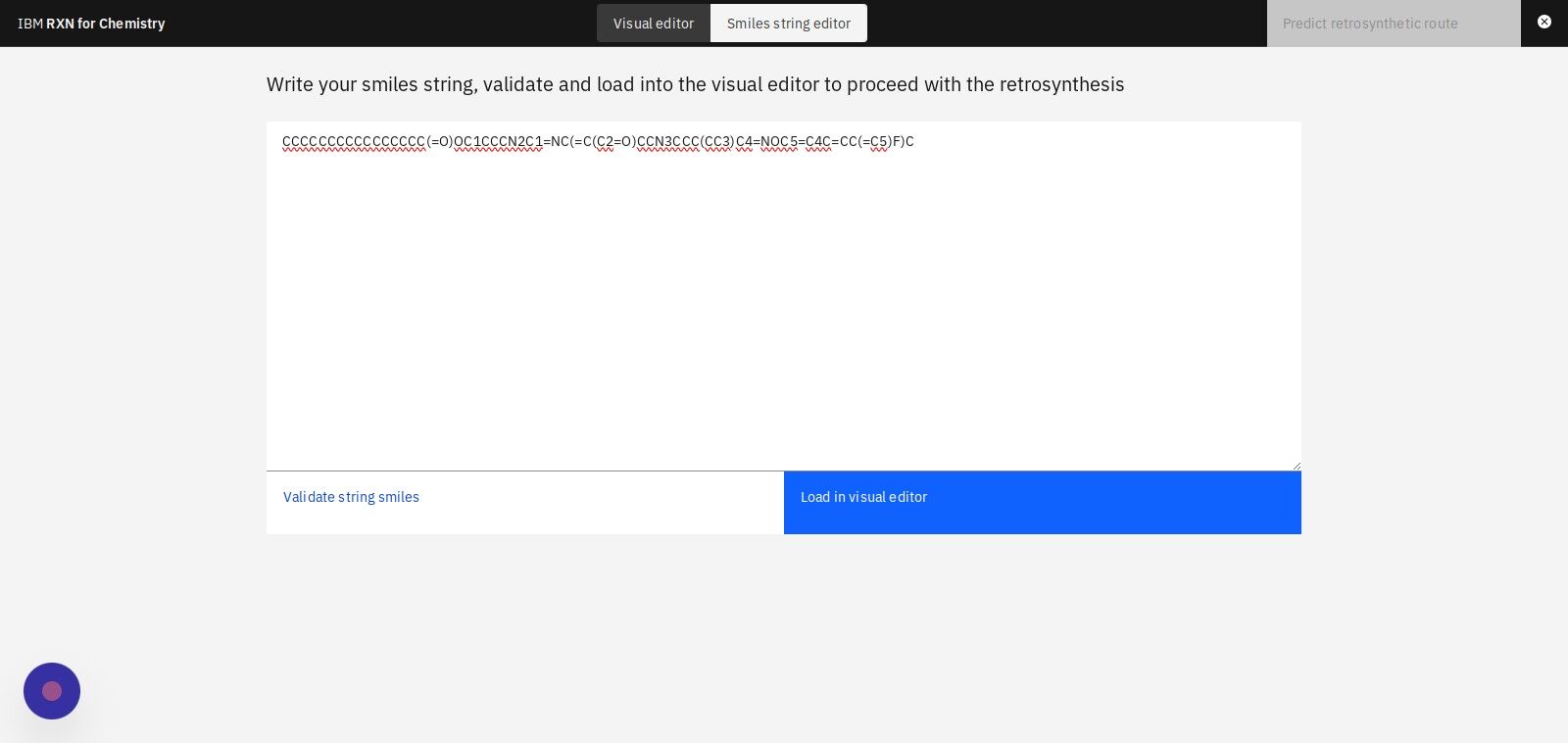
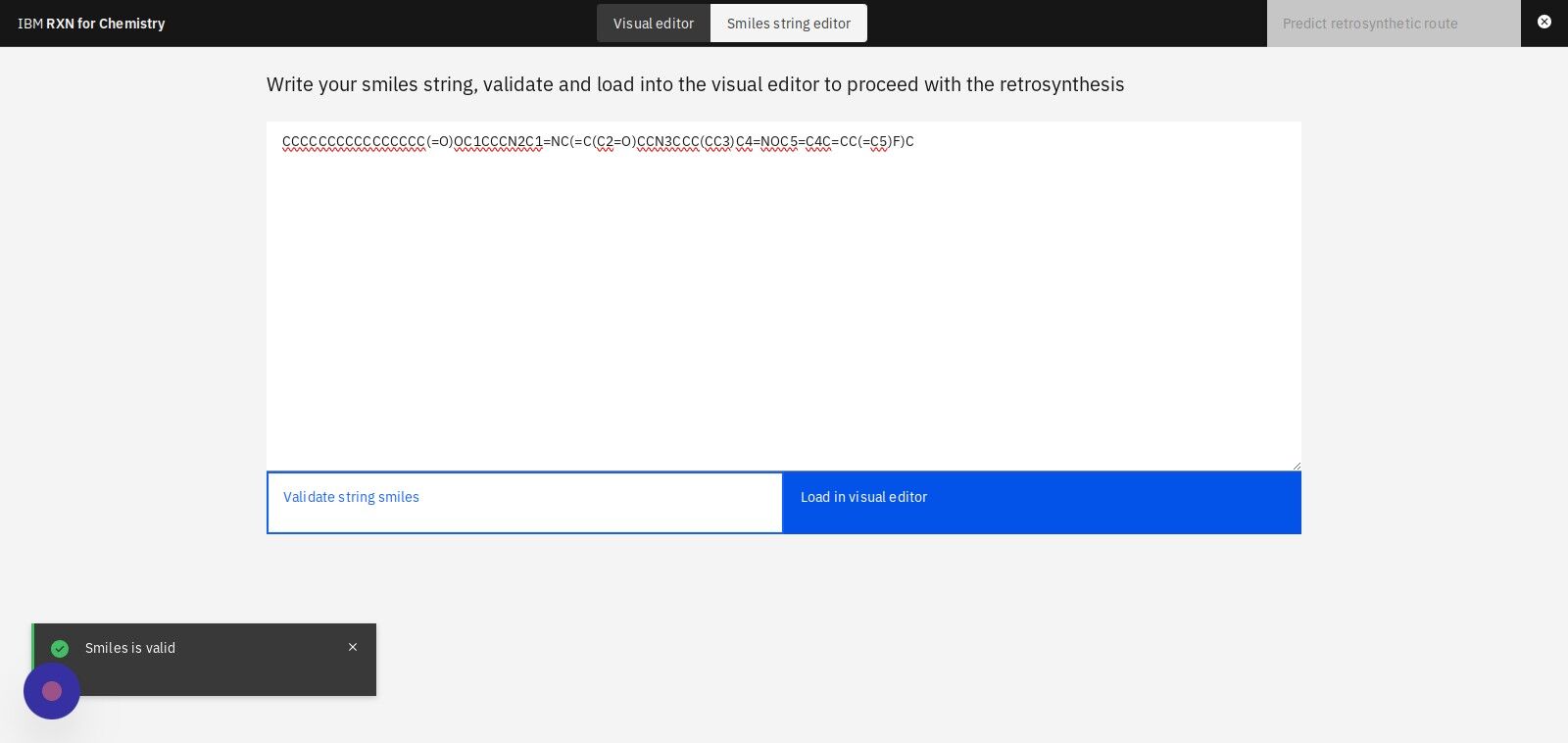
CCCCCCCCCCCCCCCC(=O)OC1CCCN2C1=NC(=C(C2=O)CCN3CCC(CC3)C4=NOC5=C4C=CC(=C5)F)C
This compound is the active ingredient in medication used to treat schizophrenia and other psychotic disorders.
Step 8: Click "Validate string smiles" to validate the structure in the SMILES string
Step 9: Click "Load in visual editor" to view a visual model of the molecule
Step 10: After viewing the molecule in the visual editor, click "Predict retrosynthetic route"
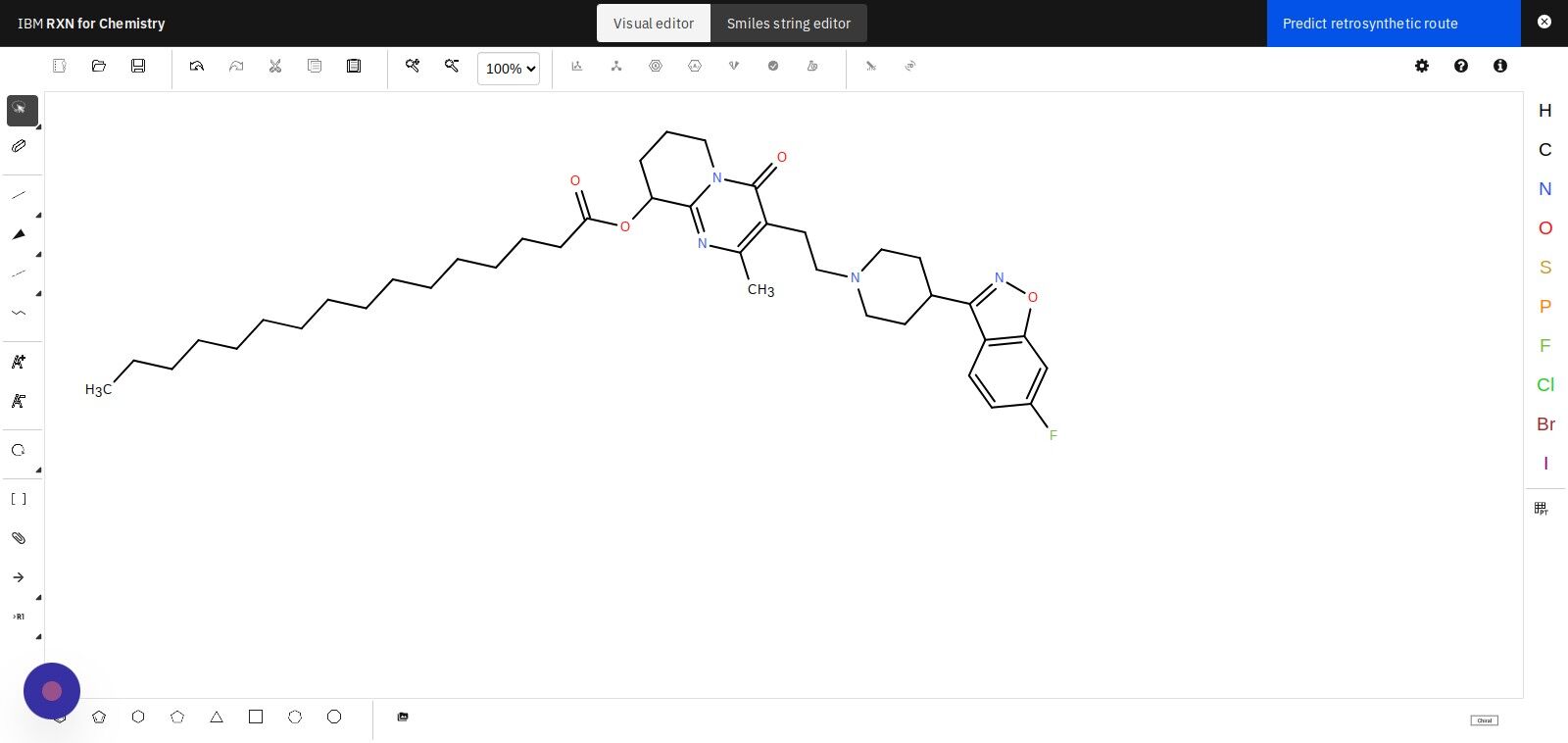
Step 11: Select "Automatic mode" in the dialog box
There is no difference in the outcome of the analysis between Automatic mode and Interactive mode, but they each provide a different way of interacting with the results.
|
Automatic mode: All of the most likely reactions and their reagents are predicted and presented in a list, allowing you to navigate and view each of the predicted retrosynthetic routes. Interactive mode: The interactive mode allows you to select step-by-step the reagents and reaction steps that you would like to use. |
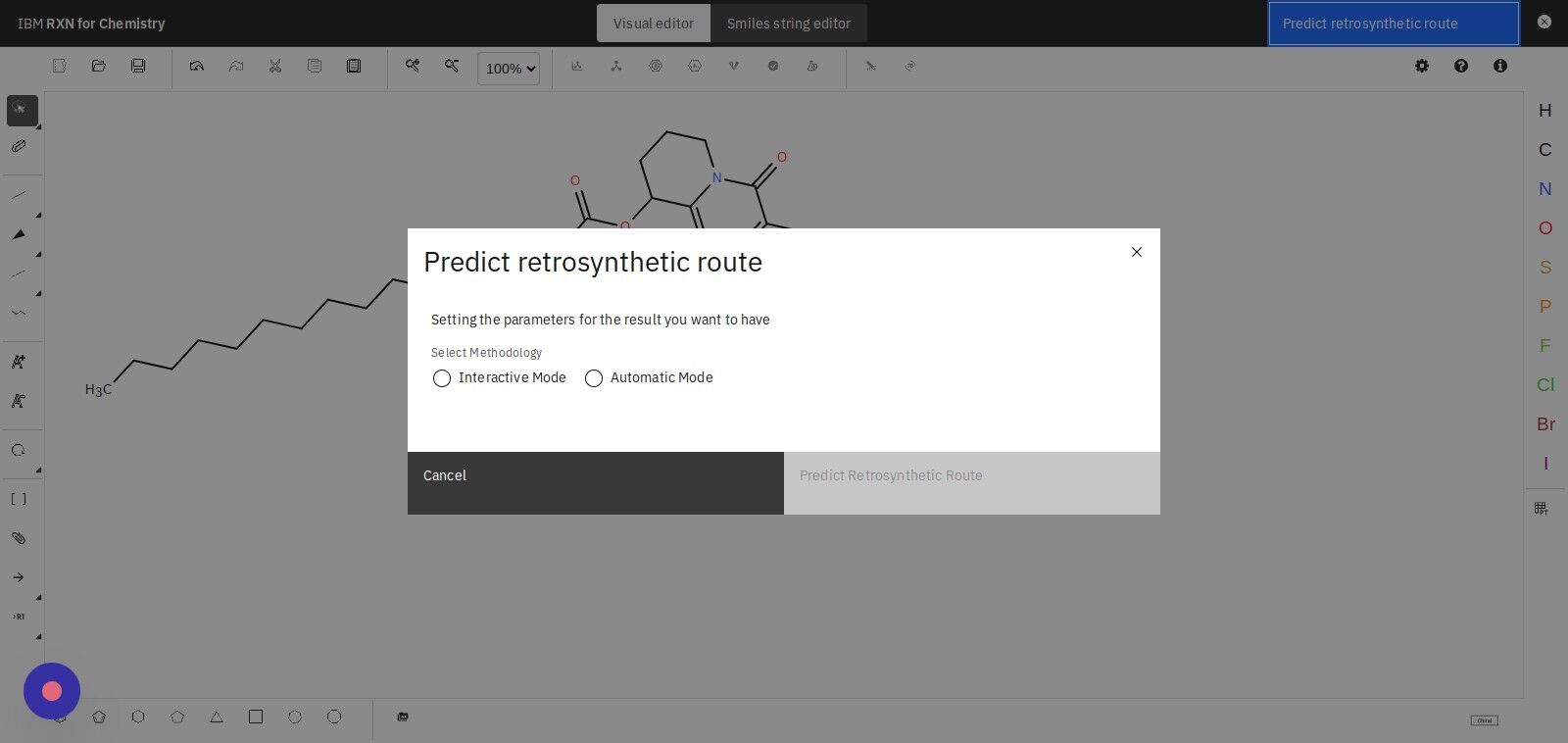
Step 12: Under "Choose AI model category" select RXN
After selecting the RXN model, select the first option under "Choose AI model version".
| AIZ and RXN are both machine learning models that have been trained on slightly different data sets. Both of these models are powerful tools for retrosynthetic analysis. |
After selecting the AI model version, you may move the slide control labeled "Speed/Quality Tuning" to set a balance between how rapidly results are completed versus how exhaustive the search is.
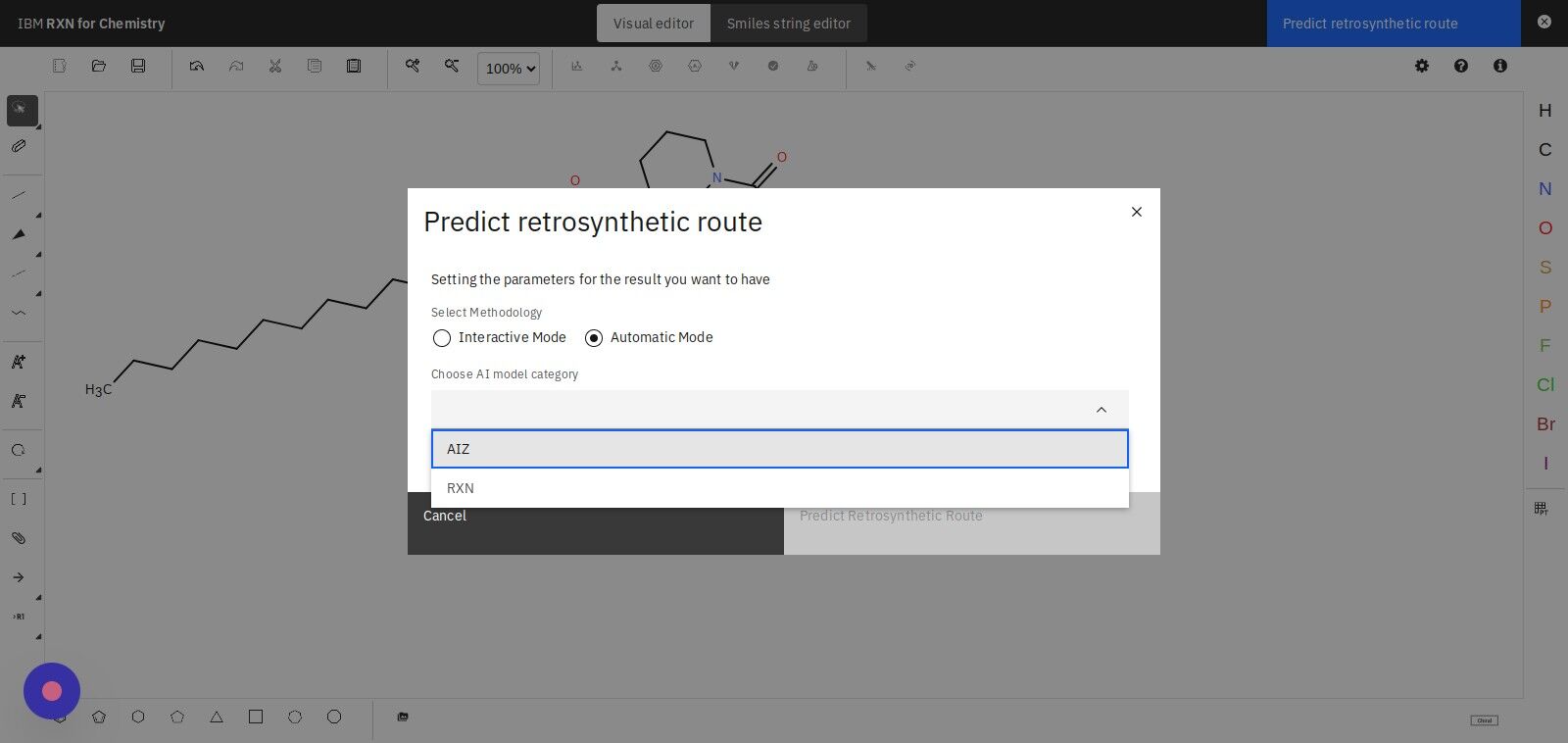
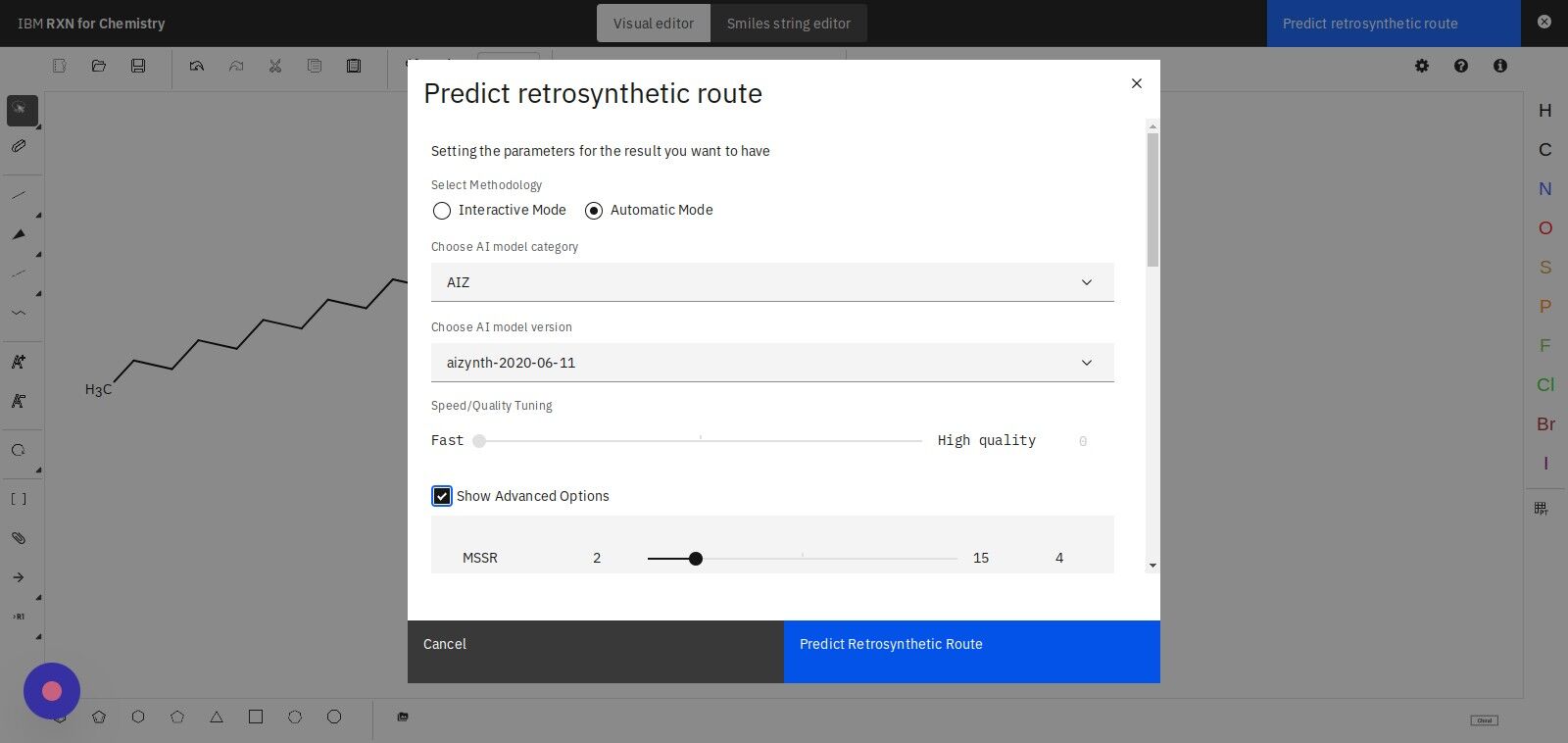
Step 13: Click the "Show Advanced Options" check box
Adjust the MSSR slide controller to a value between 2 and 15.
| MSSR is the Minimum Sum of Squared Running Digital Sum. Lower values indicate that you want only those results that more closely fit the underlying machine learning models. Higher values indicate that you want to include analyses that have a higher discrepancy between the data and machine learning models. |
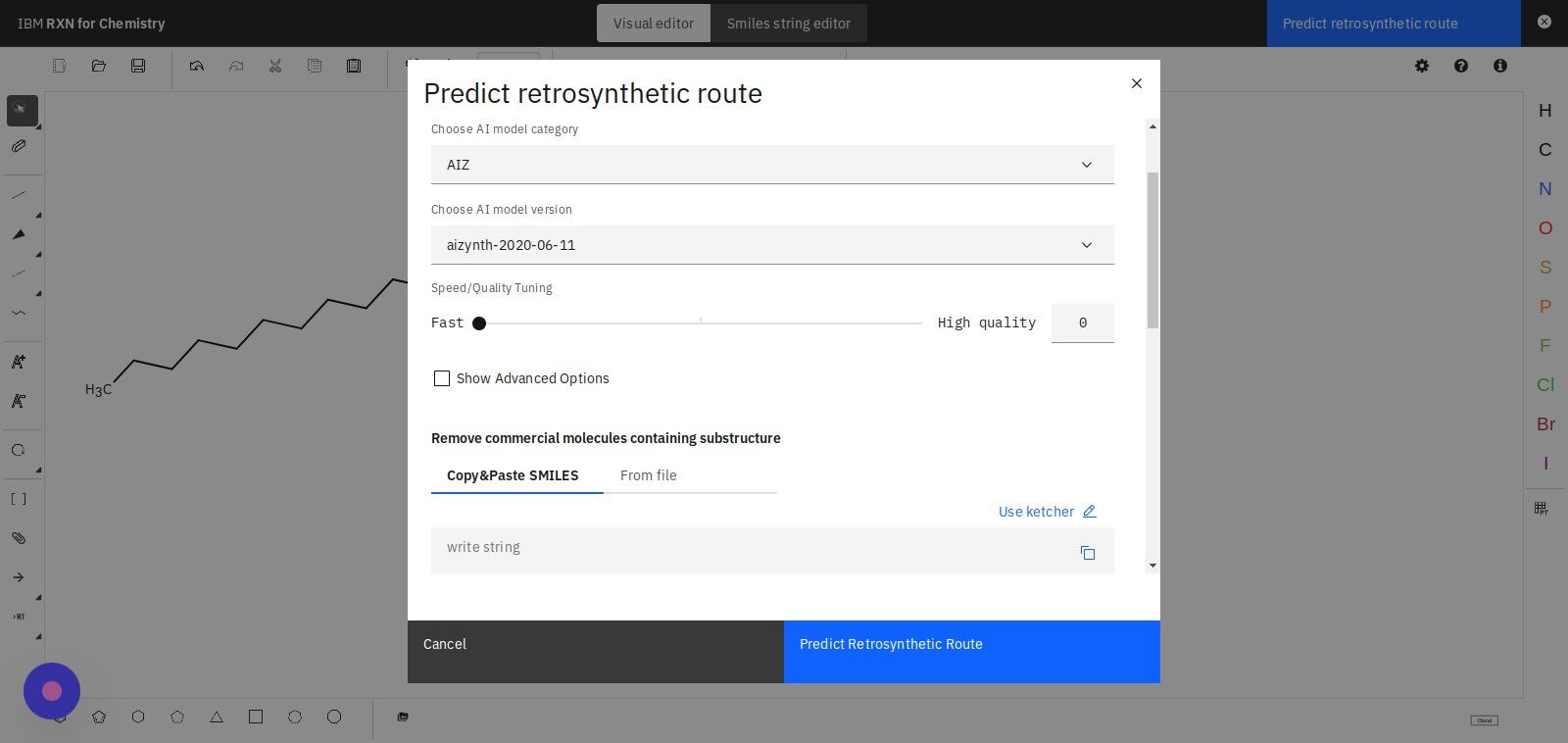
Step 14: Click "Predict Retrosynthetic Route" to begin the analysis
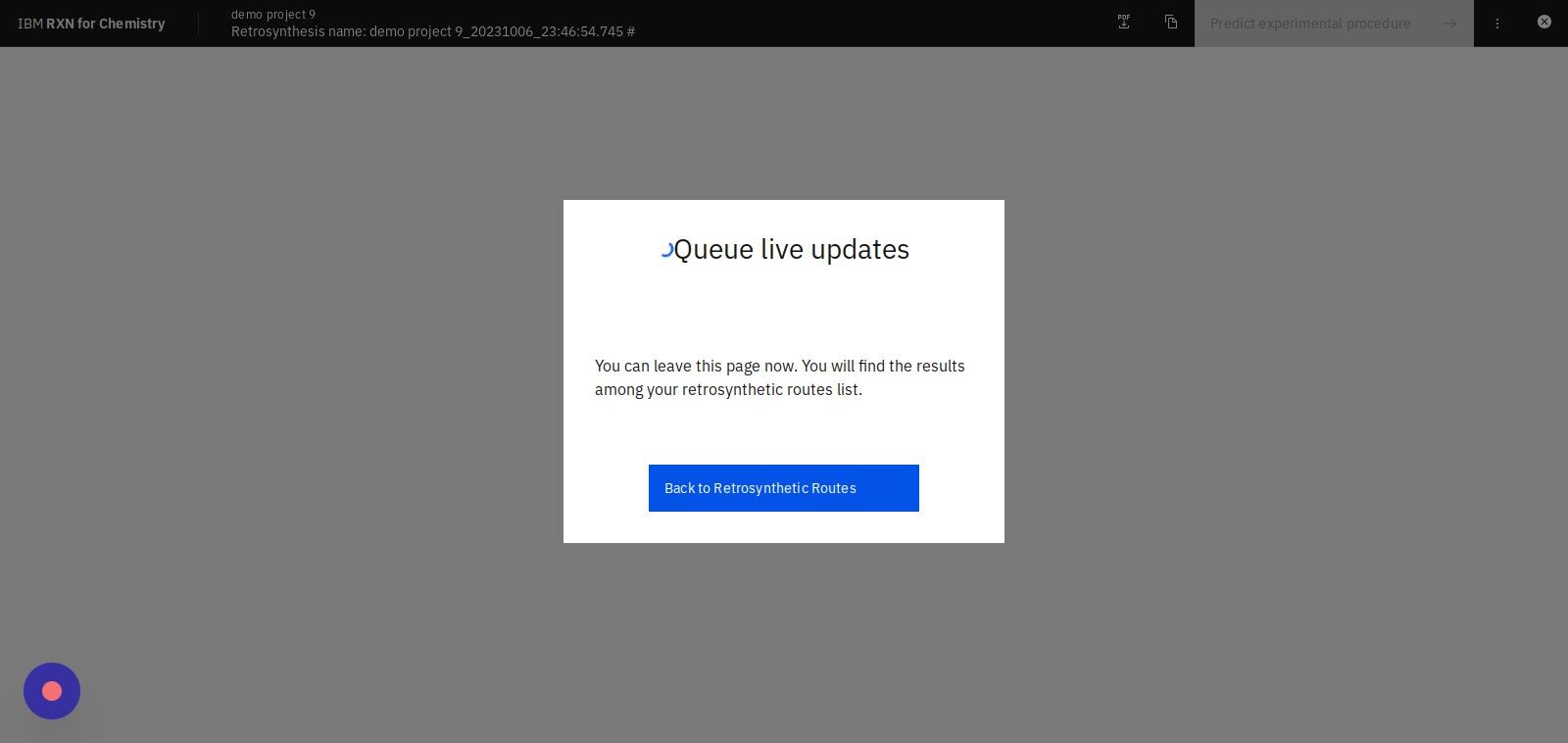
Step 15: Click "Back to Retrosynthetic Routes"
This will take you back to a page containing a listing of any previously computed retrosynthesis analyses.
| The computation of all of the retrosynthesis reactions may take some time (up to 10 minutes) depending on several factors, including (but not limited to) molecular complexity, MSSR setting, and selection of machine learning model. |
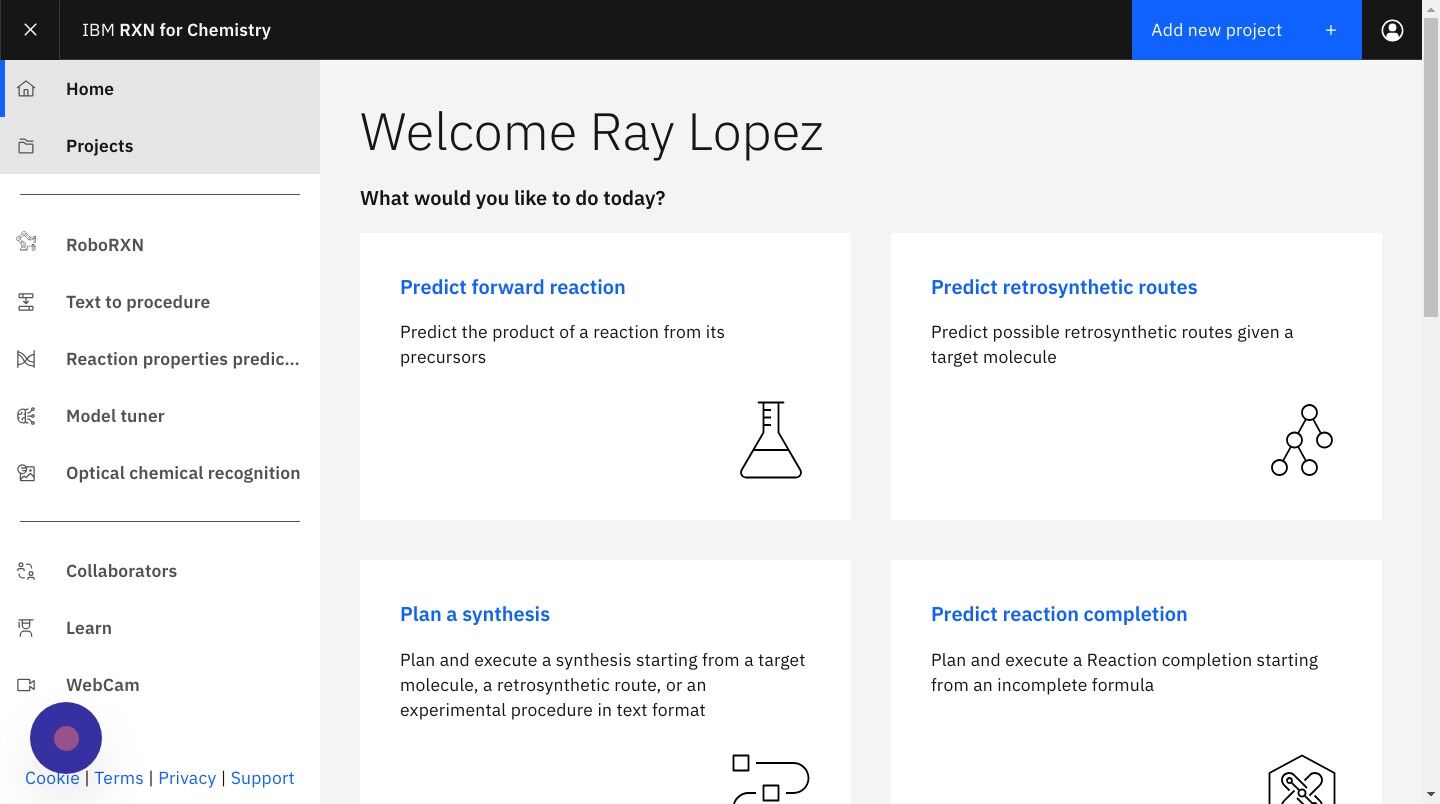
Step 16: Start from the RXN for Chemistry Home view
| The retrosynthesis computations may take several minutes. You can log off from RXN for Chemistry anytime and return to the Home page later to view the results. |
Navigate to the RXN for Chemistry Home page at https://rxn.app.accelerate.science/rxn/home.
If you are already logged into RXN for Chemisty click on the "Home" link in the left-hand sidebar.
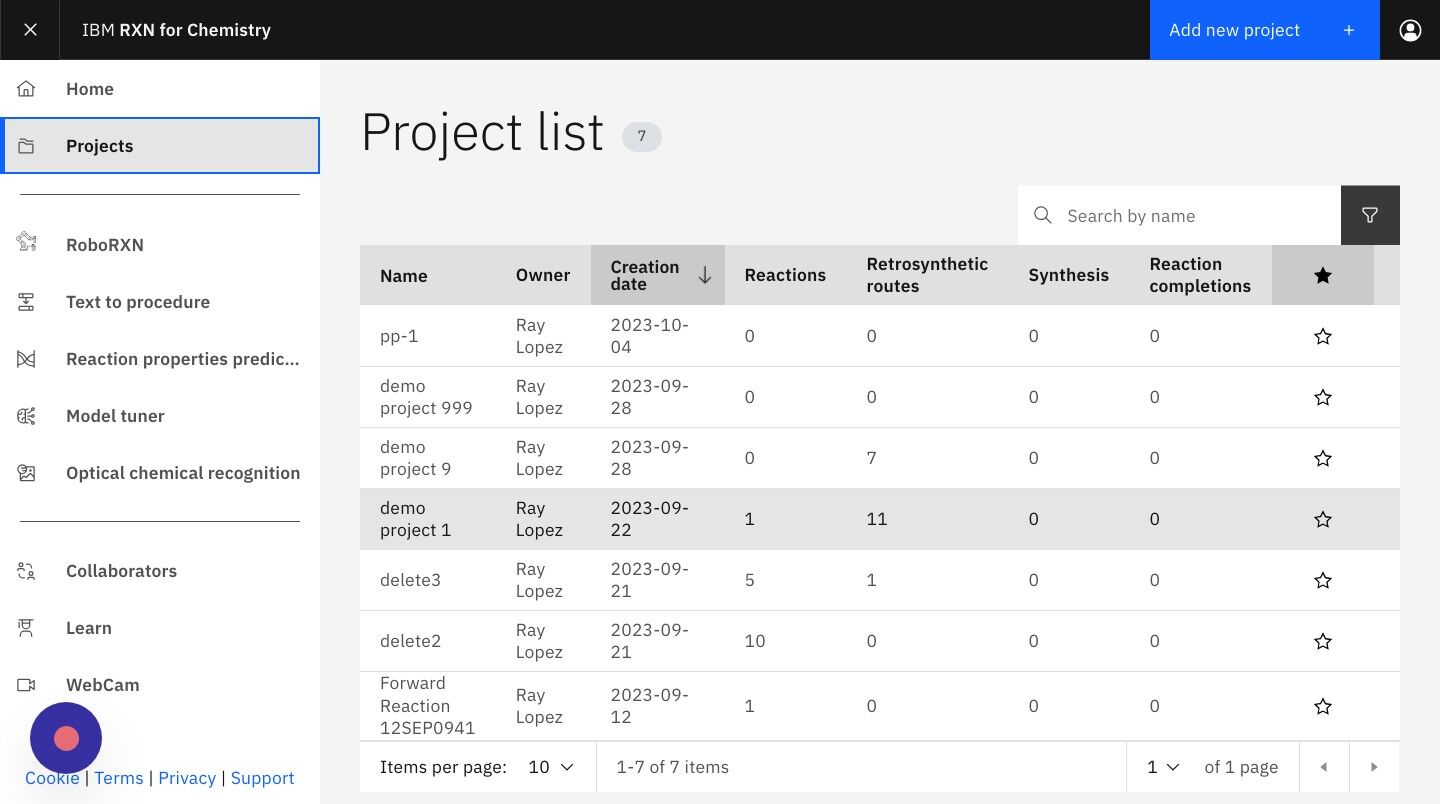
Step 17: From Home page, click "Projects"
Step 18: Click on the Project containing the retrosynthesis analysis.
Step 19: Select "AI Predictions" from the top menu, then "Retrosynthetic routes"
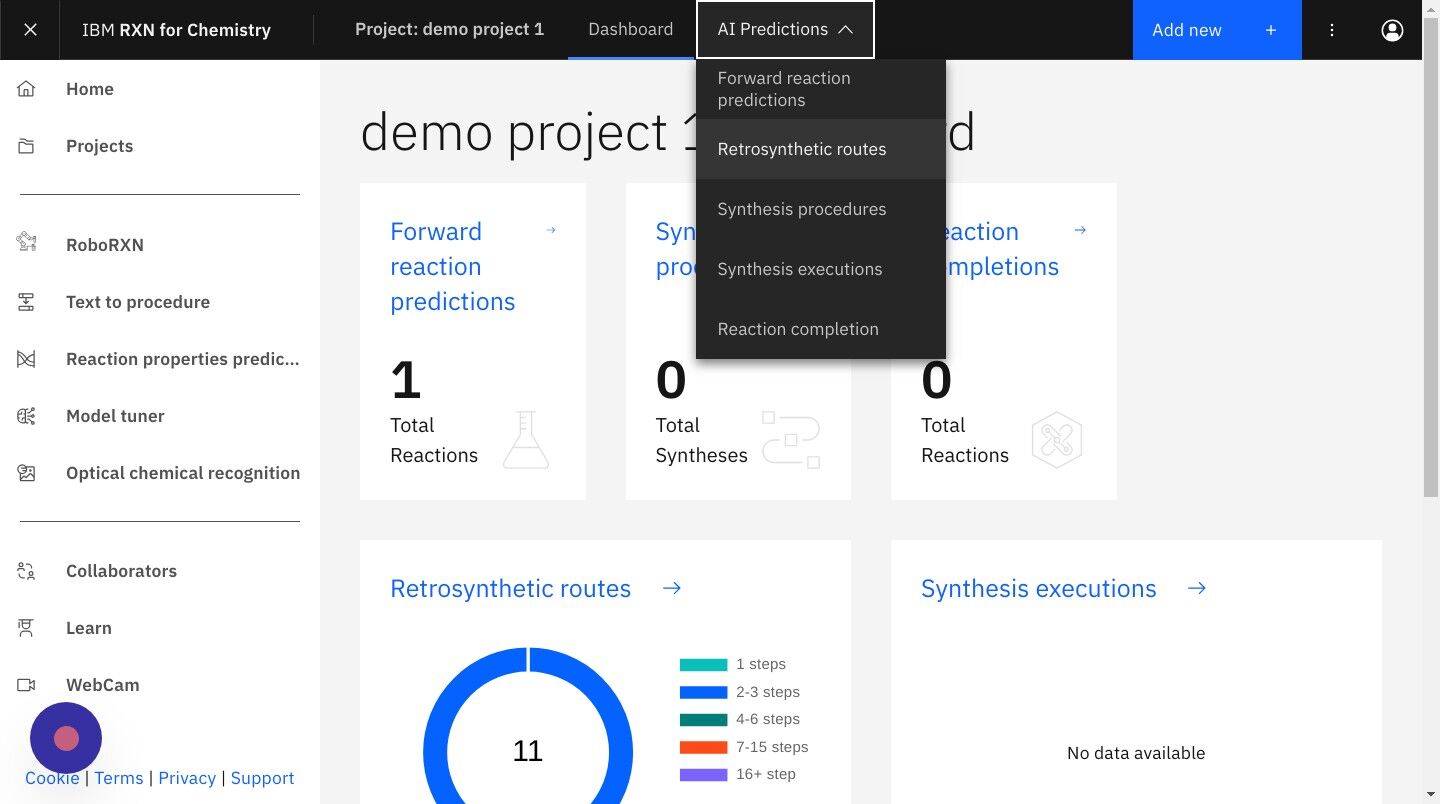
Step 20: From the Retrosynthetic routes listing select the analysis you wish to view.
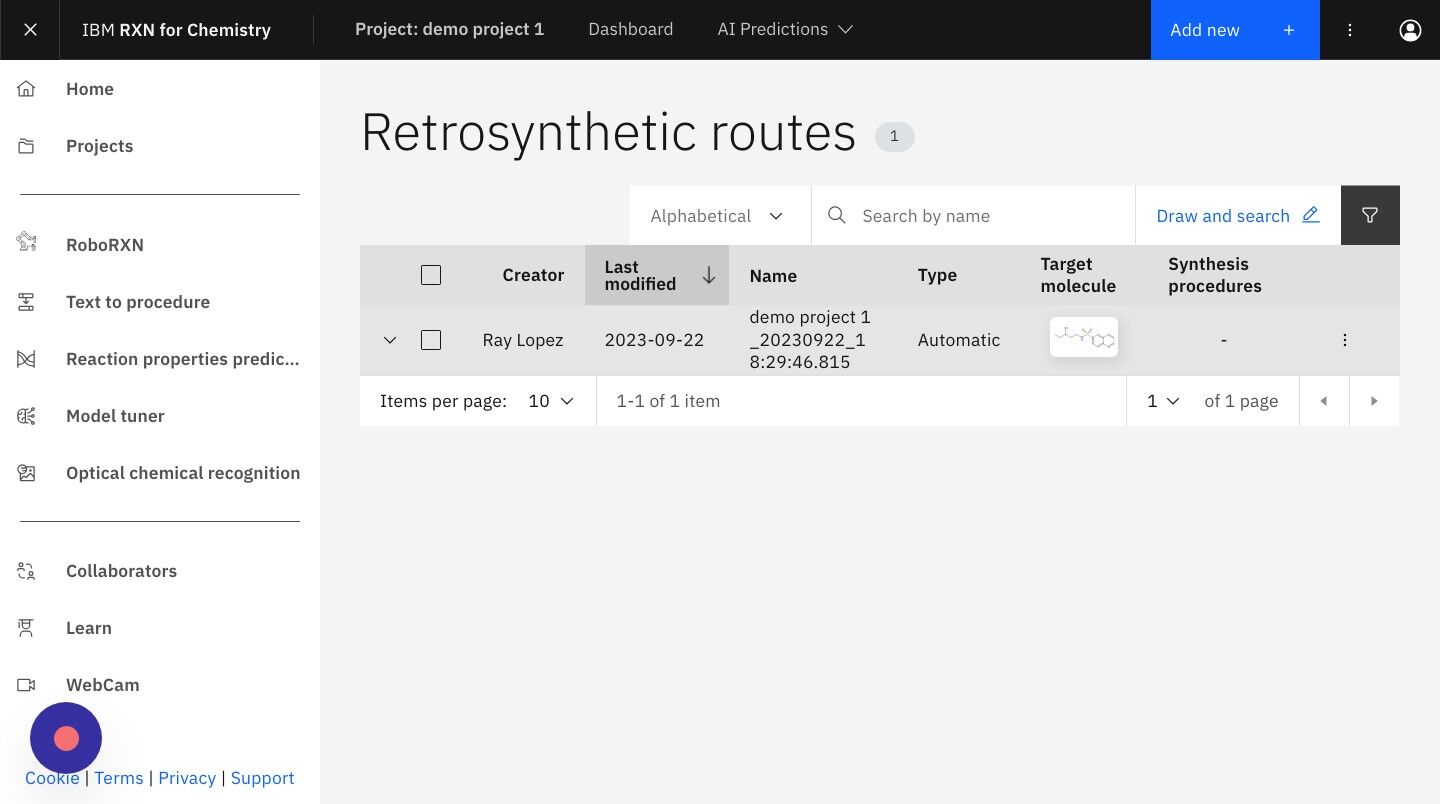
Step 21: Note the Confidence score at the top center of the page, then click on the zoom buttons for zooming in and out
|
What is confidence?
Predictions derived from machine learning models will often come with a computed confidence level. This number represents the machine’s estimate of the probability that the model’s prediction is correct, and that the predicted reaction will proceed as shown. Since it is a probability score the value of the confidence is usually given as a number between 0 and 1. Confidence scores closest to 1 are considered the best. However, the relative utility of confidence scores depends very heavily on the use case and nature of the training machine learning model. A confidence score of 0.71 might be considered unacceptable in one use case, but acceptable in another. |
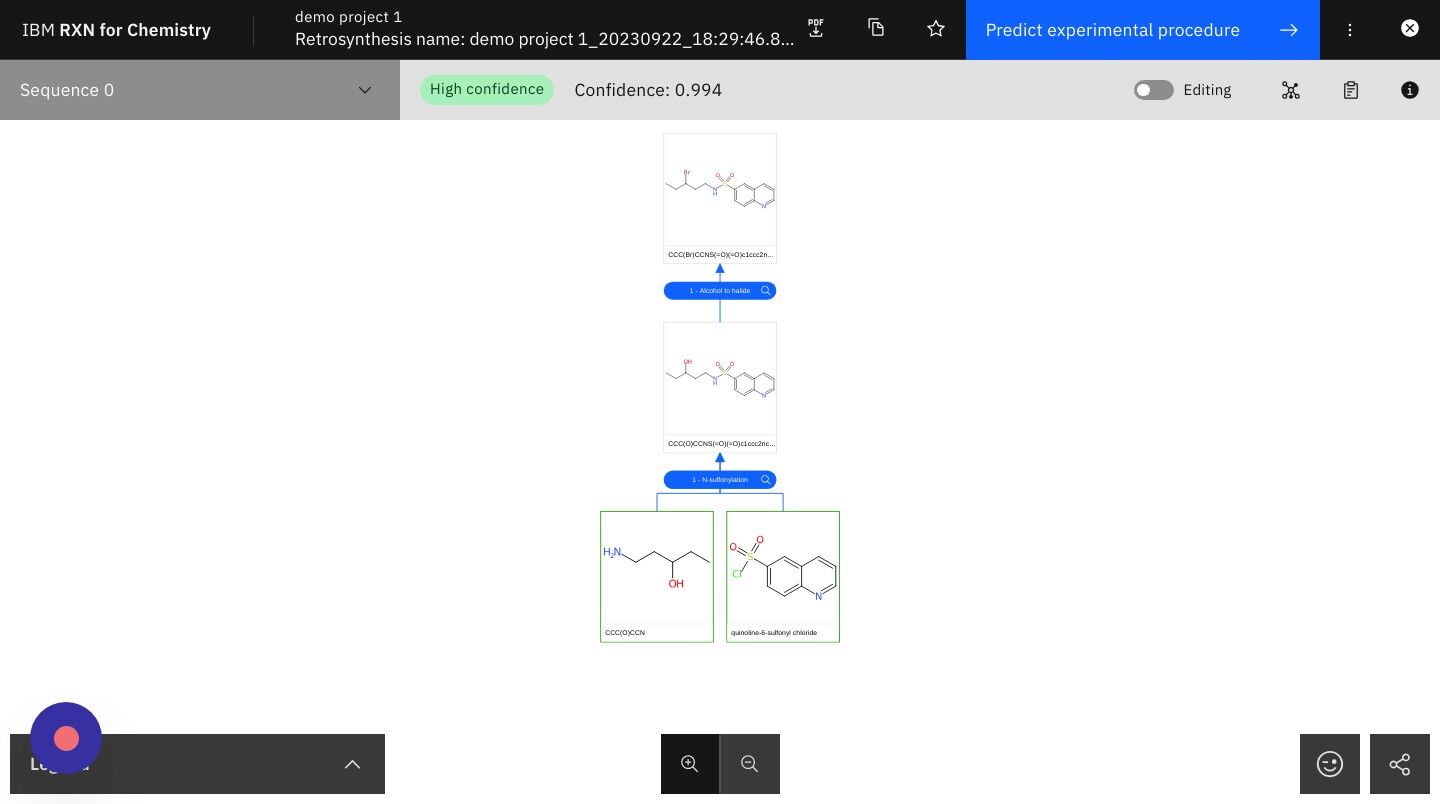
Step 22: Click on the icon shown below to view Alternative Reactions (if any) and then close Alternative Reactions sidebar
| Alternative Reactions are those reactions that may occur with lower probabiliity than the main reactions shown. |
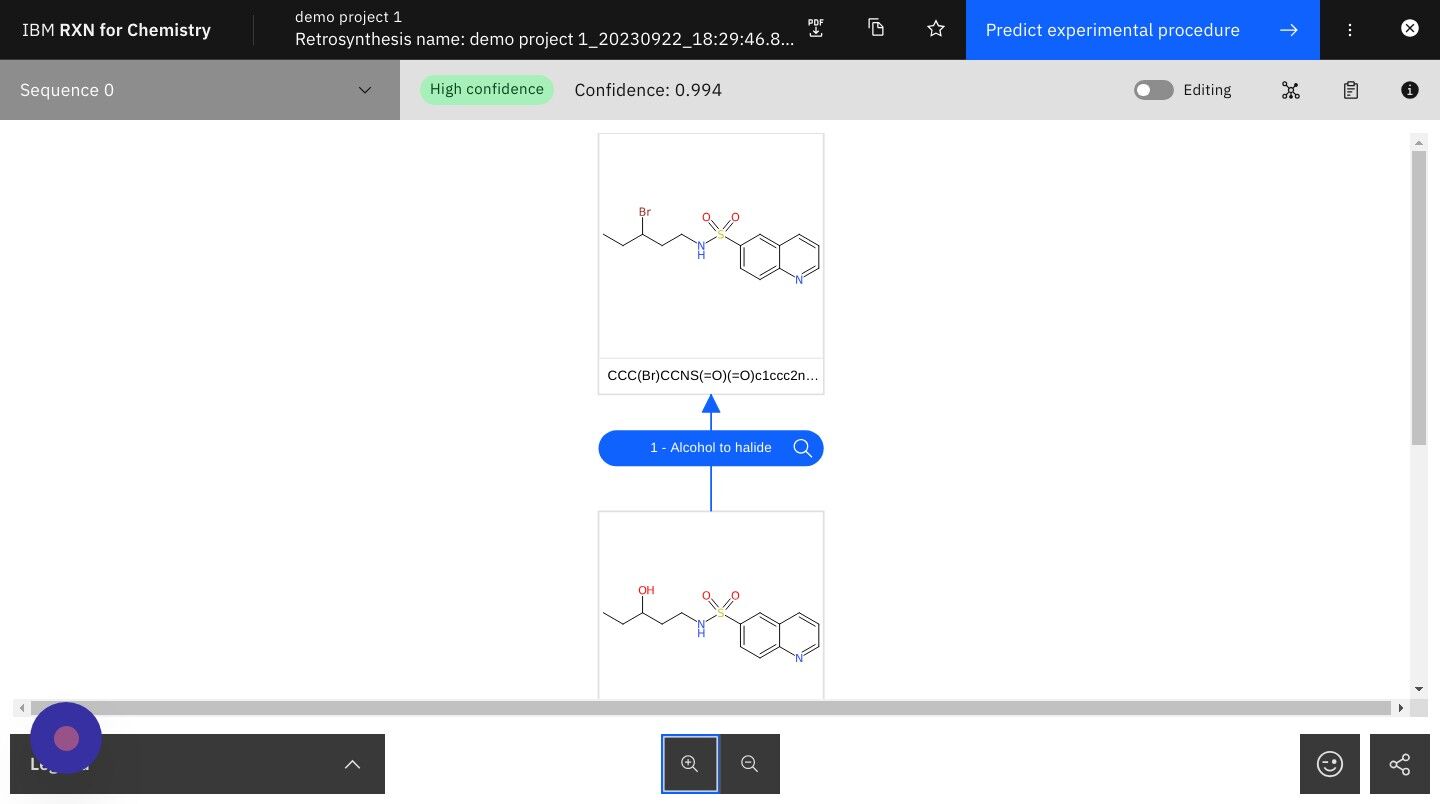
Step 23: Click on the dropdown list in the upper left of the screen
This option will allow you to view the different reaction sequences identified by RXN for Chemistry.
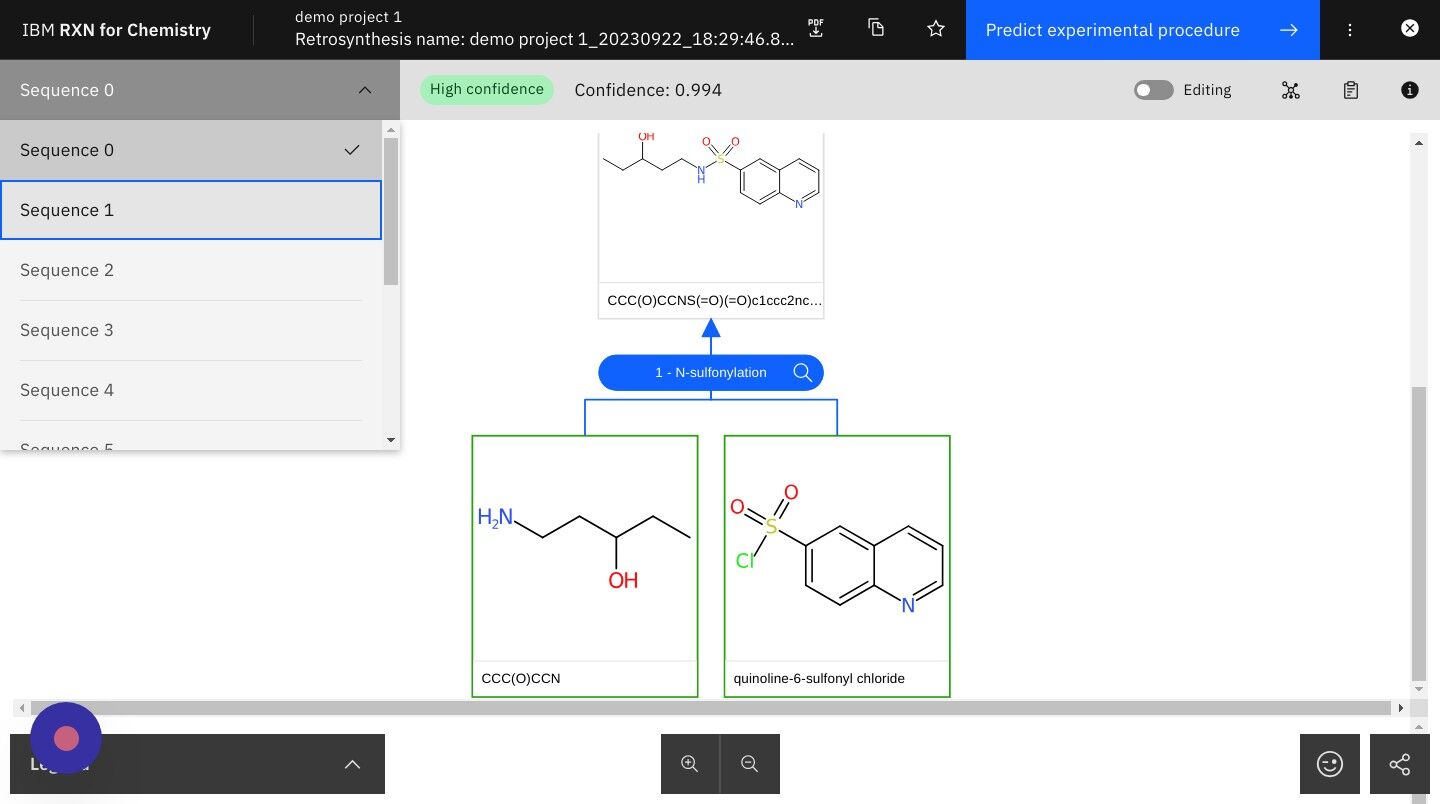
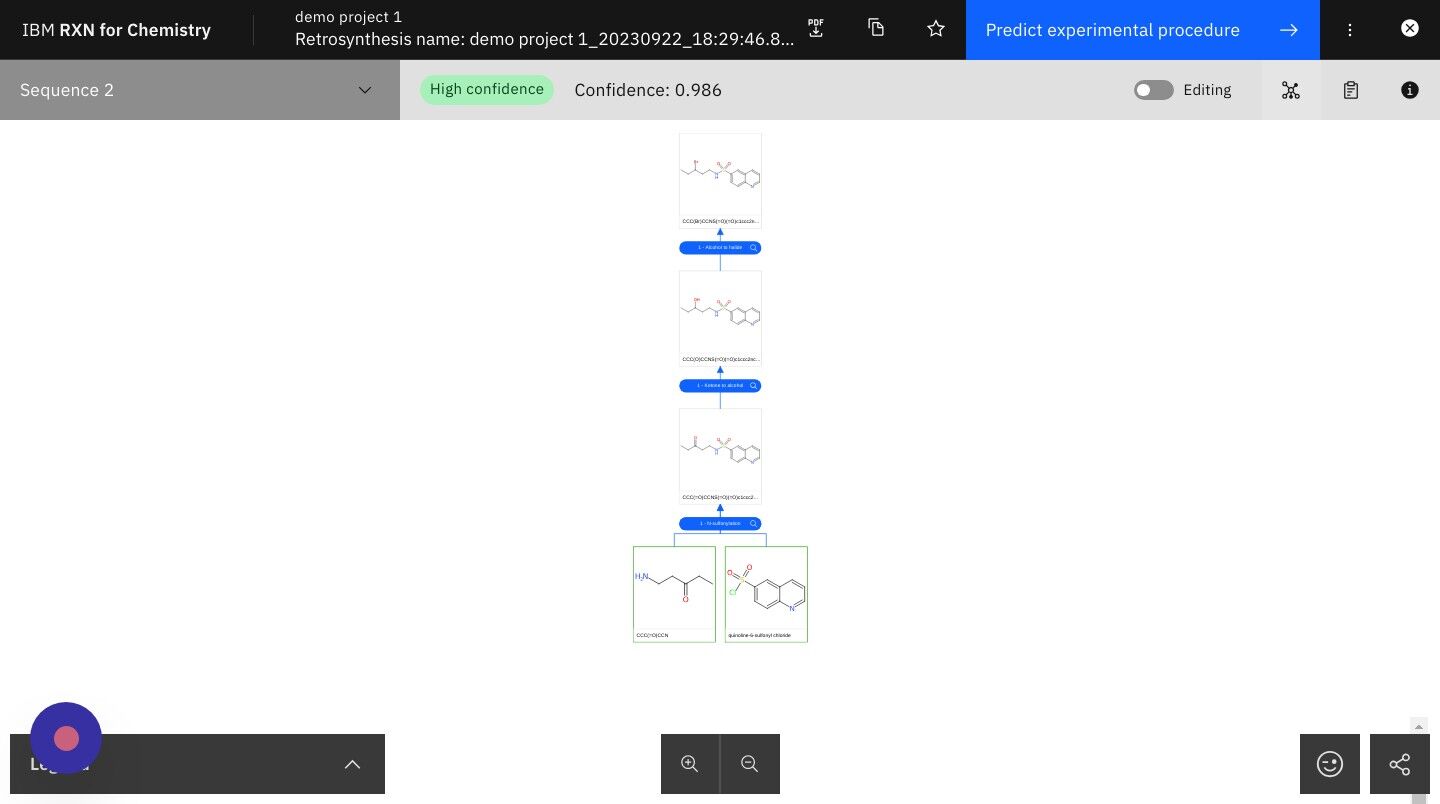
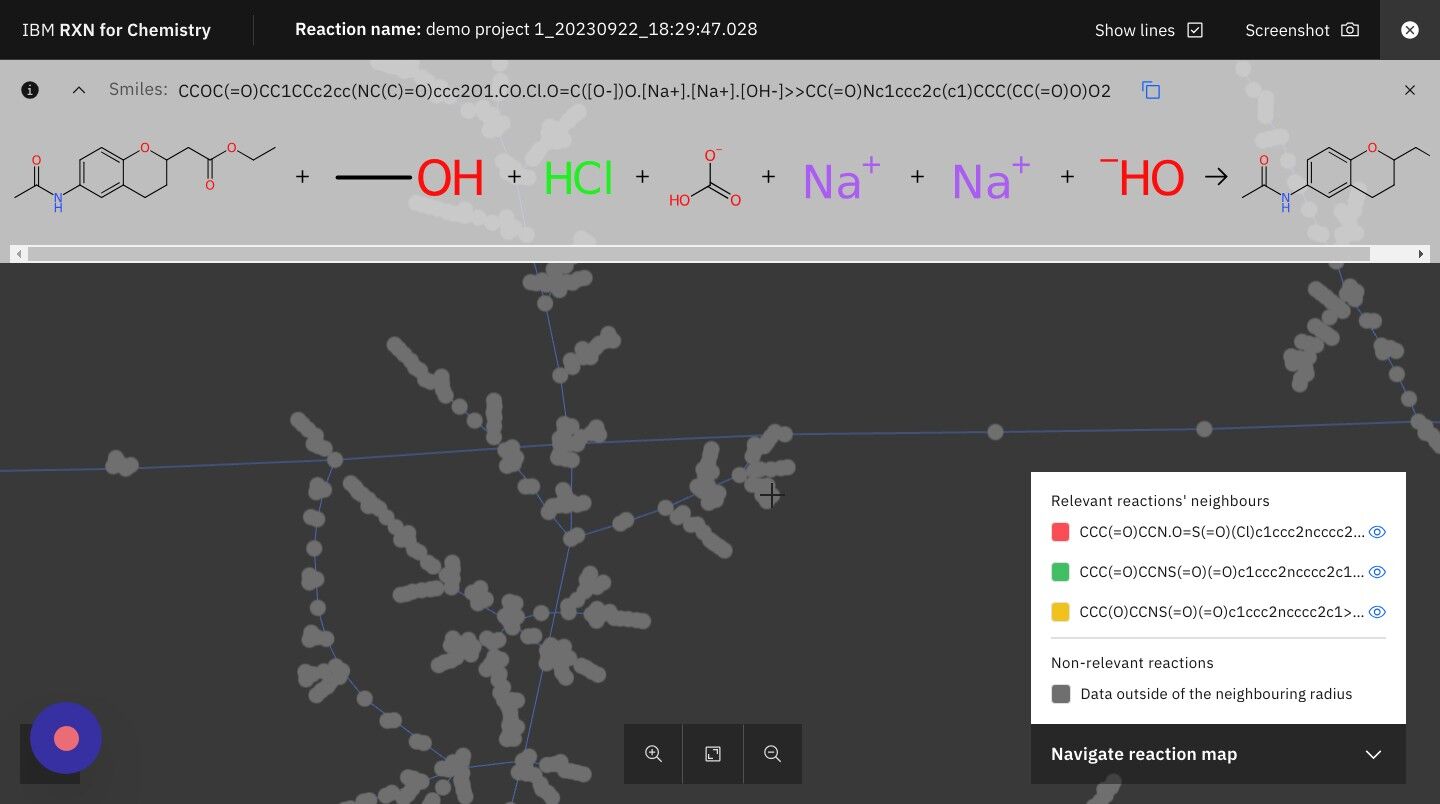
Step 24: Click on the TMAP icon
This will take you to a visual TMAP showing your retrosynthesis results in the context of other chemical structures and properties.
|
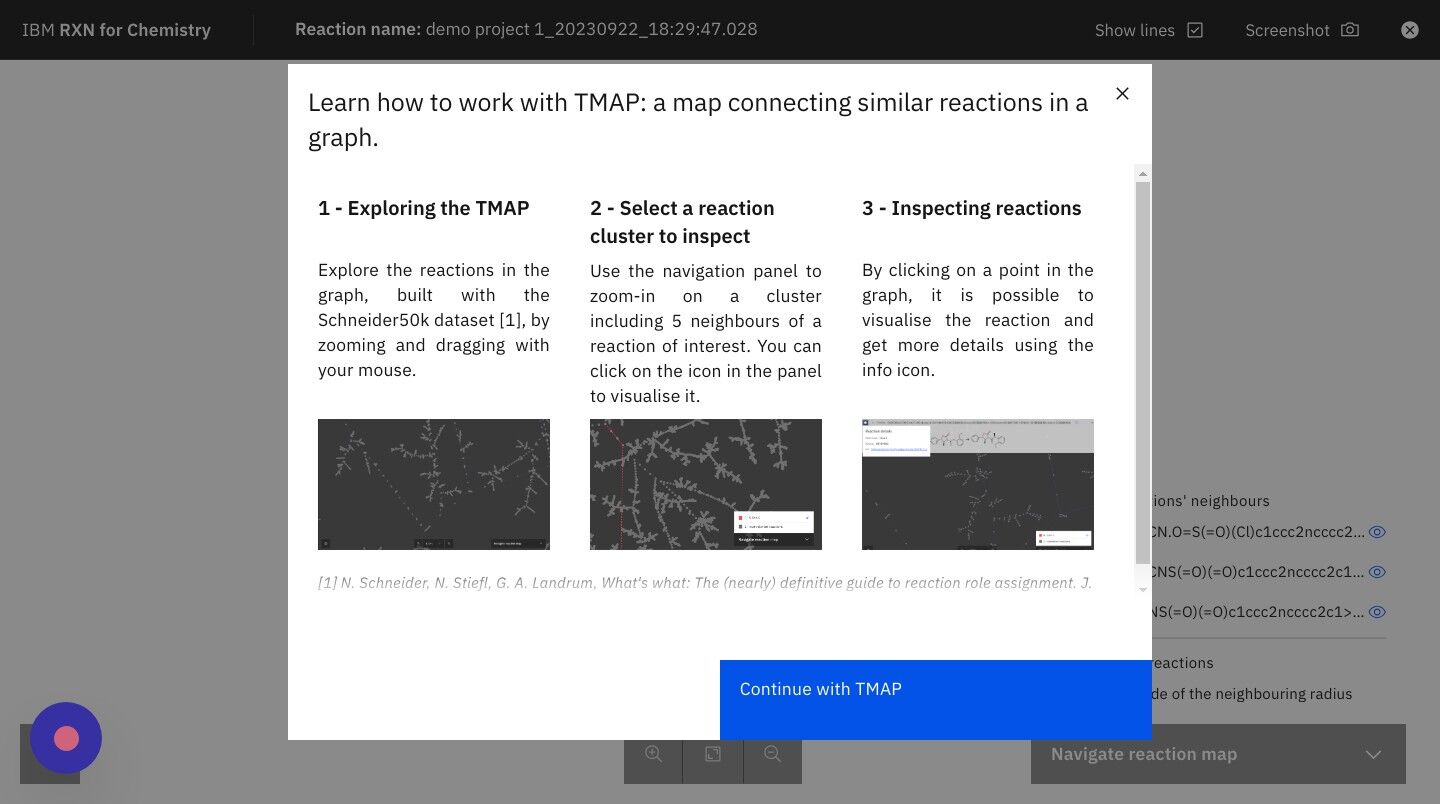
What is a TMAP? TMAP (TreeMAP) is a set of algorithms to help visualize high-dimensional data sets containing chemical structures. It visualizes such data while preserving both global and local features with a sufficient level of detail to allow for human inspection and interpretation. For more information on TMAPs, see the following: |
Step 25: After reading the TMAP guidance, click "Continue with TMAP"
Step 26: Click on the +/- zoom icons to zoom in and out of TMAP
| The legend on the right hand side of the TMAP shows you the colors used to represent your reactions most closely related neighbors in the TMAP. |
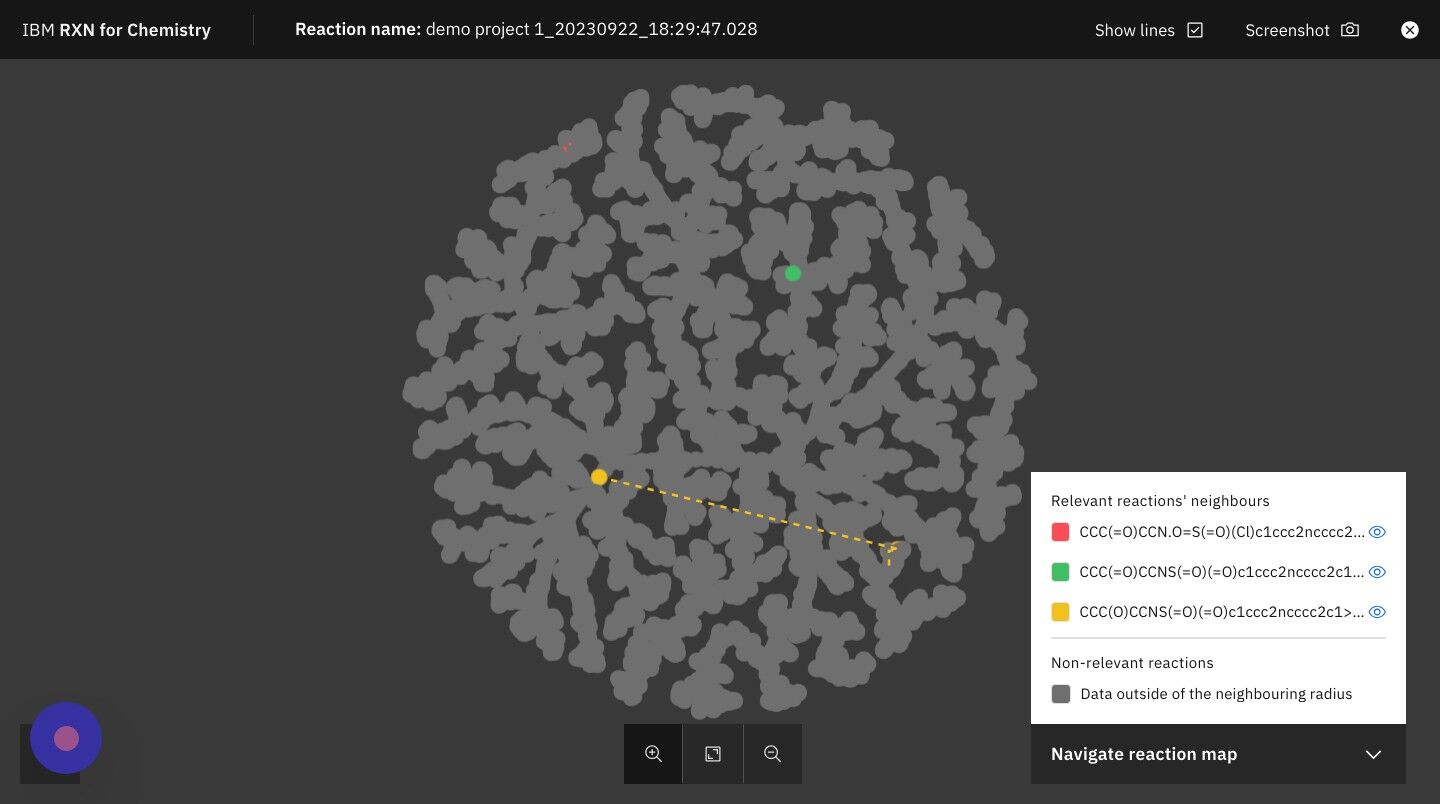
Step 27: Click on the "X" in the upper right of the screen to close the TMAP
After you have finished exploring the TMAP, close it to return to the reactions view.
Step 28: Tutorial completed
This completes the Predict Retrosynthesis tutorial for RXN for Chemistry.
In this tutorial, we:
-
Executed a retrosynthesis analysis
-
Viewed the results in a Project
-
Viewed the details of the predicted retrosynthesis reactions
-
Viewed the TreeMAP of retrosynthesis reactions
IBM®, the IBM® logo, and ibm.com® are trademarks or registered trademarks of IBM® Corporation, registered in many jurisdictions worldwide. Other product and service names might be trademarks of IBM® or other companies. A current list of IBM® trademarks is available here: Copyright and Trademark Information.